This blog post is the second of three to discuss the Genome Archive (GenArk) assembly hubs. This second post discusses examples of using the GenArk hubs’ data, with the first post about accessing the data, and the third shares technical infrastructure behind the hubs.
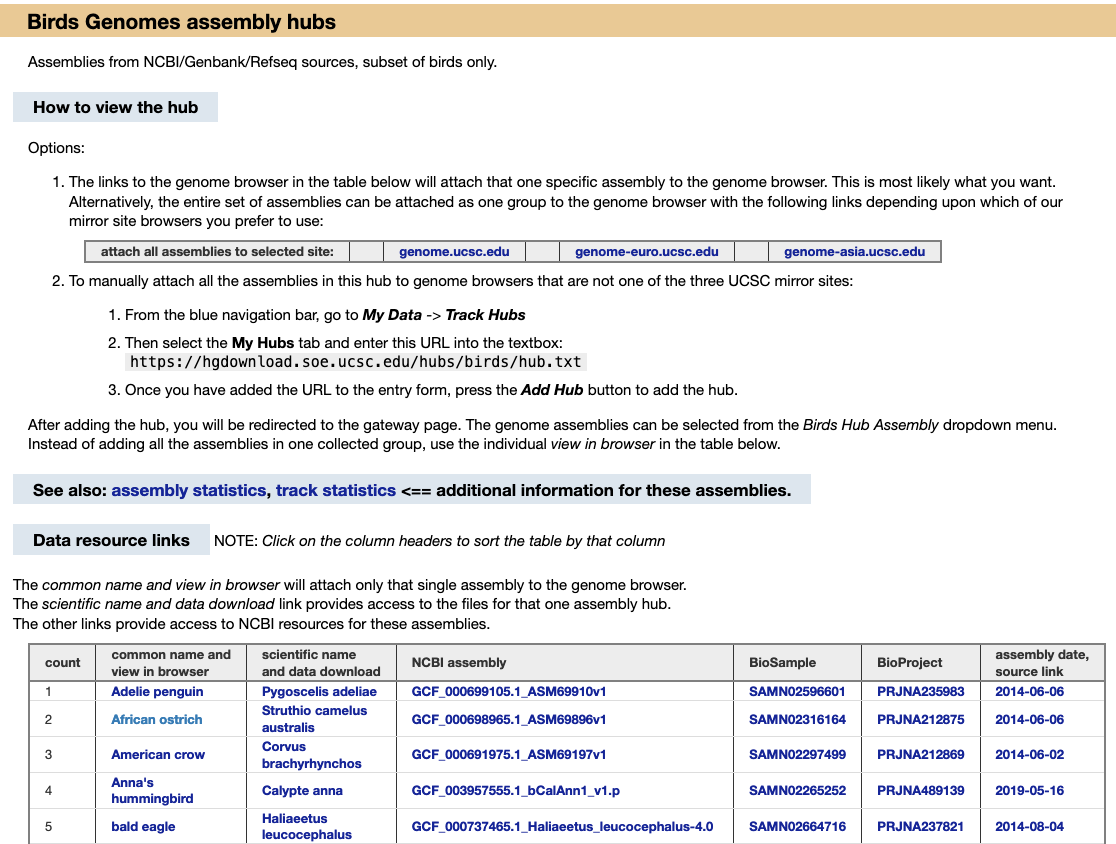
Before launching into using the new GenArk hubs, let’s go quickly over how the first blog post examined the multiple ways to access the GenArk hubs. The easiest way to find a GenArk hub is by searching the UCSC Genome Browser’s main Gateway page with a name like “hummingbird” and clicking on the GCA/GCF identifier to attach it. Another is to build direct links to NCBI GCA/GCF assembly accessions when you know them to instantly arrive at the main Browser view, such as https://genome.ucsc.edu/h/GCF_005190385.1 for narwhal. Yet another is searching the UCSC Public Hubs page or going to the main GenArk homepage where you can in turn navigate directly to individual taxonomic group pages, such as for birds.
USING THE DATA
What can you do with a GenArk Assembly hub?
The new GenArk hubs come with the ability to perform BLAT DNA queries and PCR primer searches, as well as send the genome’s DNA to external tools.
As an example, let’s say you are curious if we have a specific bat genome. The first step would be to go to the Gateway page and search “bat” and discover multiple hits.
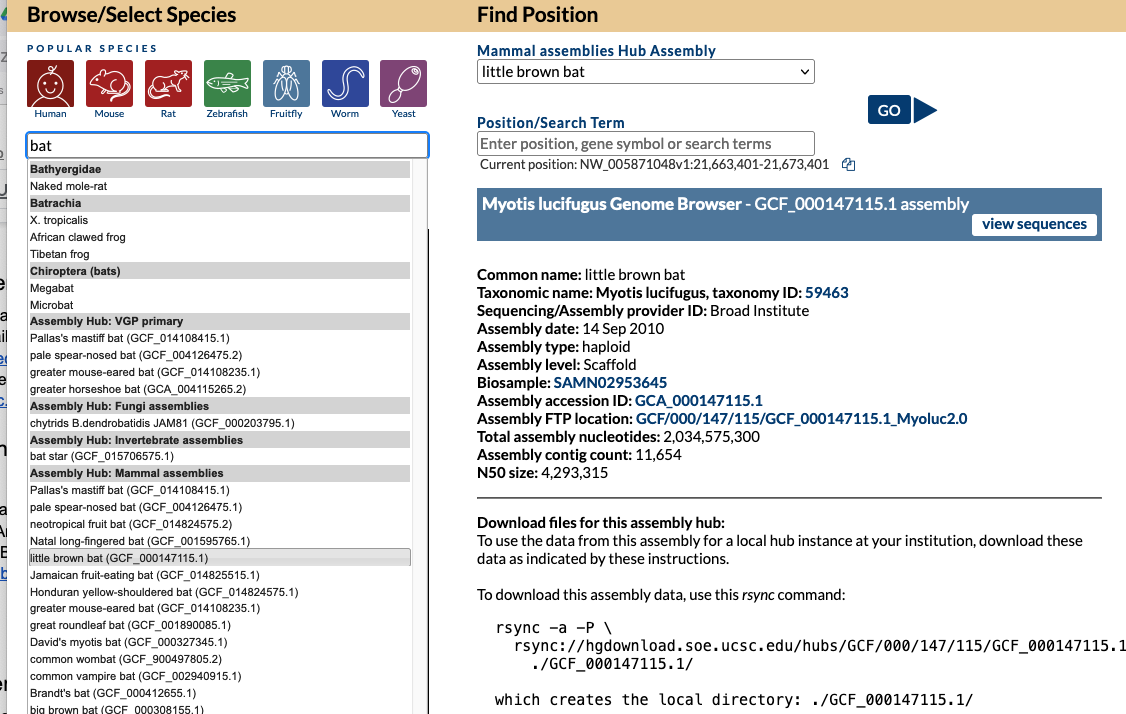
Looking at search results you see your desired specific “little brown bat” assembly and click on it so that hub is now selected, where under “Find Position” on the right there would now be “Mammal assemblies Hub Assembly” attached with “little brown bat” displayed and a specific GCF_000147115.1 NCBI accession. Clicking the “Go” button would bring you to the main Browser display. The same result happens from clicking this short direct GenArk /h/ hub link: https://genome.ucsc.edu/h/GCF_000147115.1
BLAT DNA Search
With this bat genome displaying if you had a short DNA sequence you wanted to search, you could paste it right in the top search box. For instance, after clicking the above link, try pasting on the main browser display CATTAGGCAAATATATGCATATAAGTTCTTTGTTTAATCTCT and hit “go”. The result, shown after a few seconds, will be sequence matches across the little brown bat genome. You can also go to the top Tools menu and then select “Blat”, and do the same step of pasting DNA sequence, required when searching especially longer strings. The Blat Tool page also allows you to search alternative sequences.
BLAT Protein Search
On the Tools > Blat page you can put in protein sequences to search. This is especially interesting if you want to find the location of a known protein from another species in your genome of interest. For example, if again you are on the little brown bat genome and you go to the Blat page, try to blat this portion of the human SOD1 protein:, LSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGN
You will find a match (again note for protein searches, be sure to go to the Tools > Blat page). When viewing the results you can either click a “browser” link to see the matching spots on the genome. Or if you click a “details” link you will see the side-by-side alignment like this image below.

Besides DNA and protein searches, BLAT also allows translated RNA and translated DNA searches. Also the results from BLAT searches can be saved as custom tracks. This allows you to download and save these annotations, or save them in Sessions making the results more permanent and shareable. See this other blog post about sharing sessions for more information: https://bit.ly/UCSC_blog_sharing
PCR Primer Search
GenArk also provides PCR primer searches, by going to the Tools menu and selecting PCR. With the same “little brown bat” genome loaded, for instance, go to the Tools menu and select “In-Silico PCR” to arrive on the PCR page. Then enter these two primers, forward primer: AGTCATGGTCTCAGGAACCG and reverse primer: GTTACTAGGGCTCAGACCTC (there is no need in this example to click any other settings).
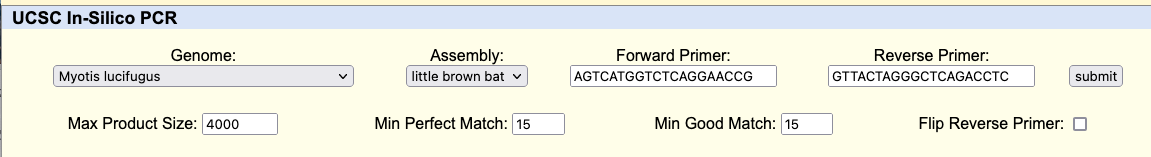
Then click “submit” to search the “little brown bat” genome for matches.
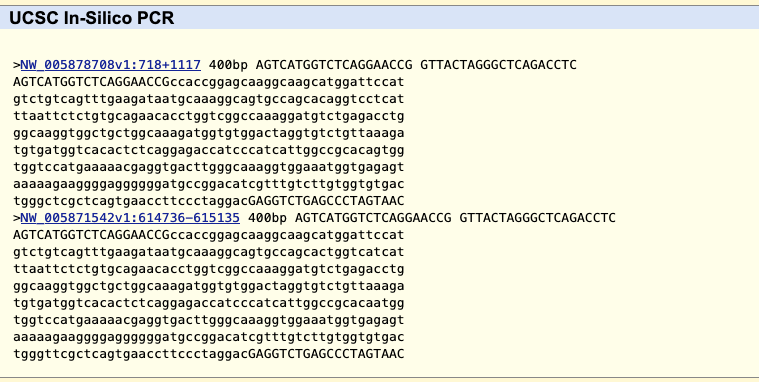
The results will be two hits, in part because this assembly has 11,654 scaffolds with some identical sequences (to see all the scaffolds click the “view sequences” link on the Gateway page described later).
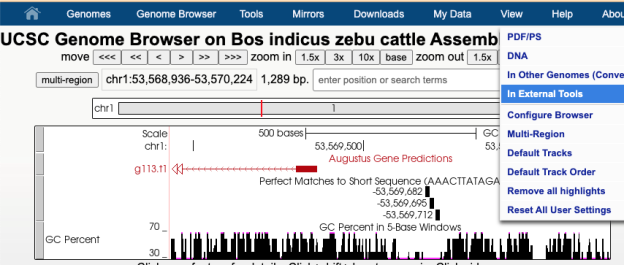
Send DNA to External Tools
Another way to use GenArk hubs is to send the current DNA in the viewing window to external sites. By going to the View menu you can select the “In External Tools” option and export the current DNA for processing outside of the UCSC Genome Browser.
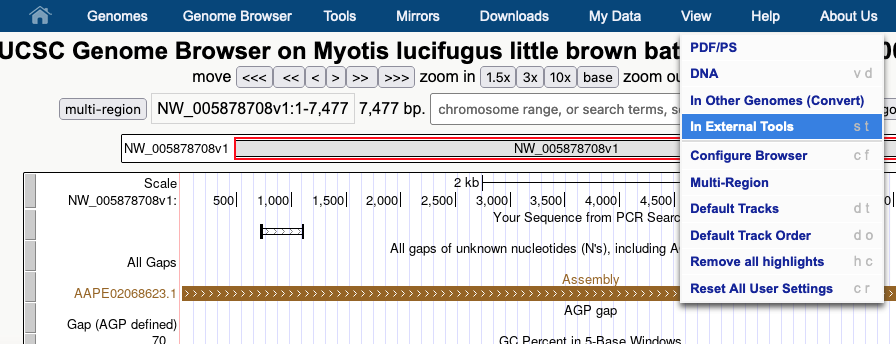

In this case all of the options are presented as available for this 7,477 bp span, except for RNAfold, which requires the viewer to zoom in to less than 5 kpb, before sending the DNA to that external tool.
Send DNA to External Tools -Primer Design: Primer-BLAST
If you were interested in PCR Primer design in this region you could use the Primer3Plus or Primer-BLAST links. The Primer-BLAST link starts a job at NCBI, where after some time the results at NCBI will be optimal PCR Primers for this stretch of DNA. Here are example results sending the 7,477 bp span of the NW_005878708v1 little brown bat scaffold to NCBI.
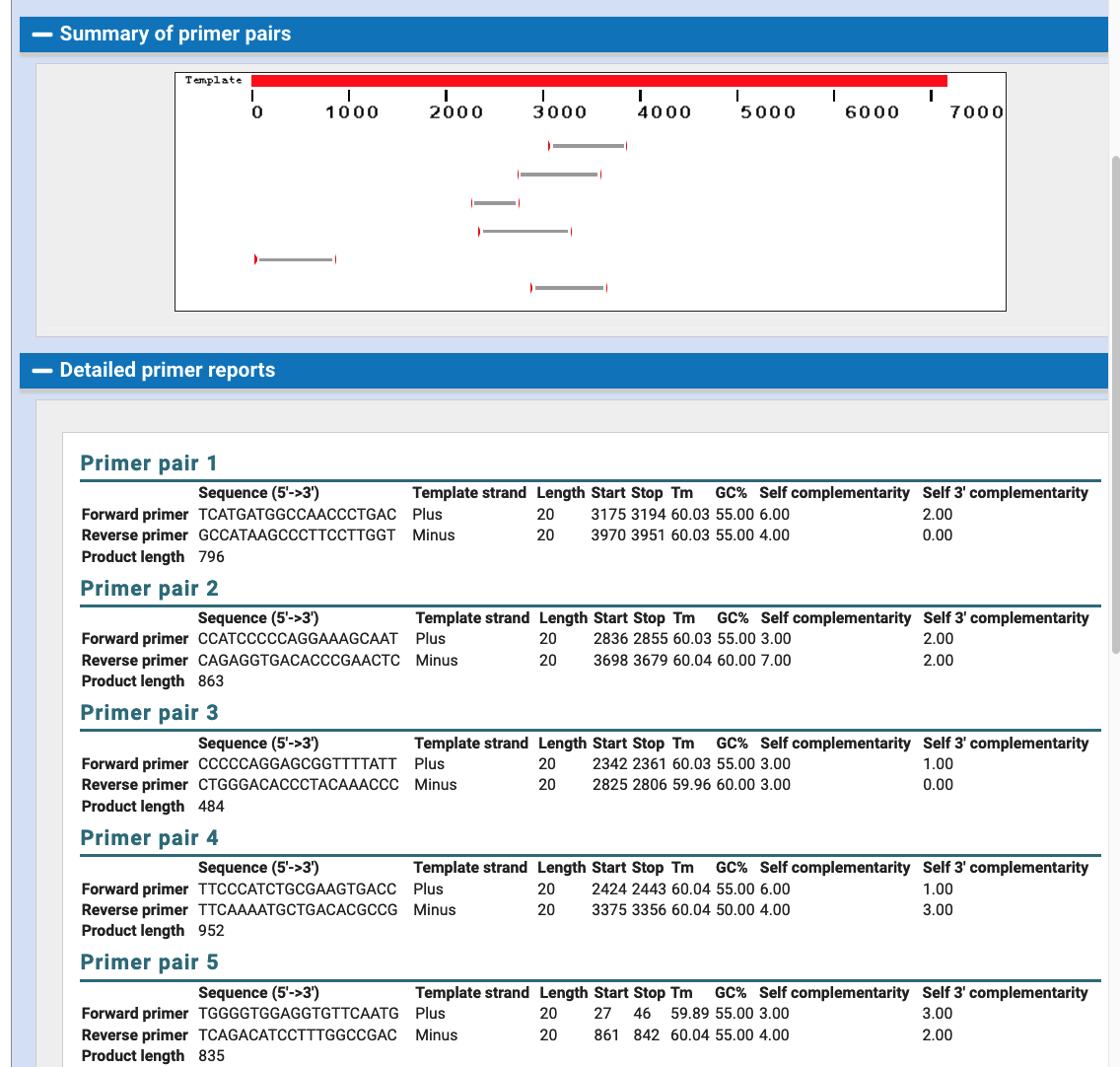
With these results, one can return to the UCSC PCR Tool to test each result in order to discover if these primers will have potential off-target results beyond the desired chromosome.
Send DNA to External Tools -Primer Design: Primer3Plus
Another PCR Primer design option in the “In External Tools” menu is Primer3Plus. Here are example results sending the same 7,477 bp span of the NW_005878708v1 little brown bat scaffold to Primer3Plus.
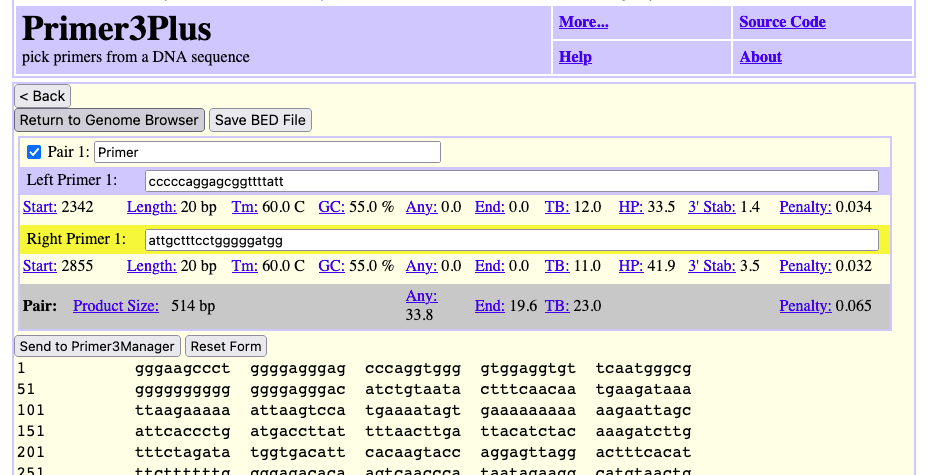
Primer3Plus has the added benefits of a “Return to Genome Browser” button (top left) that if clicked will dynamically generate a custom track of the results to be seen back on the UCSC Genome Browser.

Above the Primer3Plus custom track identifies the input region that was sent (top grey bar), and then the individual left and right matching primer pair locations. At UCSC the primers can then be tested again with the UCSC PCR Tool where a highlight for the Primer3Plus suggested “Primer 5” is highlighted in the above image.
Send DNA to External Tools for oligo-analysis
Another tool you can export DNA of interest to is Regulatory Sequence Analysis Tools (RSAT) Metazoa for motif discovery. For instance, when looking at a GenArk assembly for Zebu Cattle, https://genome.ucsc.edu/h/GCF_000247795.1, using the View menu and In External Tools option one could select the RSAT link. RSAT provides a way to analyze the DNA sequence for transcription factor binding sites and over-represented oligo-nucleotides. Because RSAT requests your organism, in this example Bos taurus was used as a relative to zebu cattle, allowing for proceeding to request examination of the region. The DNA being sent in this example was near a region for the start of a gene predicted by Augustus. One of the RSAT results was a predicted motif, aaacttatagata, just upstream of the transcription start site for the predicted gene.
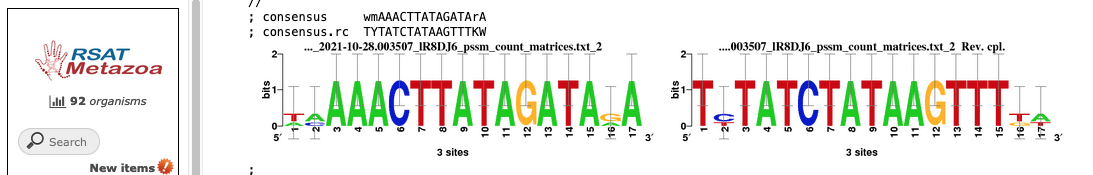
By going back to the UCSC Genome Browser and clicking into the Short Match track (under the top Mapping section) and pasting in the motif sequence, aaacttatagata, a display in the GenArk hub of where these matches occurred could be visualized.
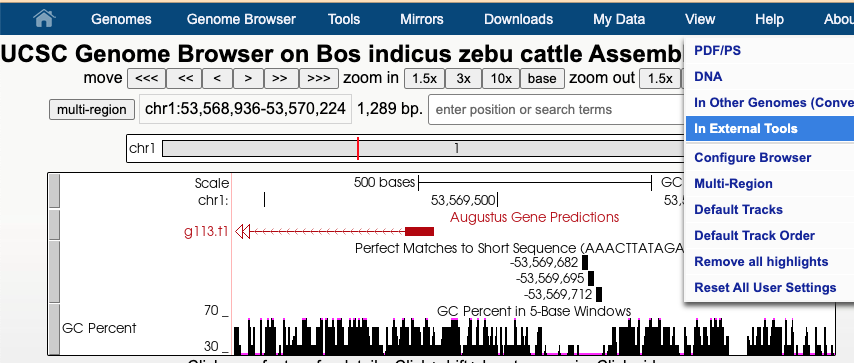
The Short Match track’s ability to visualize the motif identifies the potential binding sites of transcription factors, predicted by RSAT. This Browser view of the Zebu Cattle GenArk assembly hub can be viewed with this Public Session link.
Can I add custom tracks to a GenArk Assembly hub?
Yes, users can add tracks to their data by going to the My Data menu and then selecting Custom Tracks to paste in information. Simple text-based tracks can be loaded, or more complicated binary-indexed files such as BAMs or VCFs or bigBeds can be loaded as well.
How do I name my sequences for my custom tracks?
Another special feature of GenArk hubs is that they are loaded with a special chromAlias file allowing for multiple alias names. When building custom tracks the scaffold names for sequences need to match the names in the assembly, but many options exist. For instance, with the Zebu Cattle genome, https://genome.ucsc.edu/h/GCF_000247795.1, if you type “v s” to view sequences, or click the top “Genomes” name and then the “view sequences” button, you will end up on a page where all the scaffolds of a genome are displayed.

Scrolling down you on the resulting page you will see a link titled “GCF_000247795.1.chromAlias.txt” which will have results like this:
# sequenceName alias names assembly: GCF_000247795.1_Bos_indicus_1.0
chr1 1 CM003021.1 NC_032650.1
chr10 10 CM003030.1 NC_032659.1
chr11 11 CM003031.1 NC_032660.1
...What this chromAlias.txt file displays is how “chr1”, or “1”, or “CM003021.1” or “NC_032650.1” can be used to create custom tracks on chromosome one for this assembly (i.e., BED custom tracks “chr1 300 500” = “1 300 500” = “CM003021.1 300 500” = “NC_032650.1 300 500”).
Can I add a Track Hub to a GenArk Assembly Hub?
Yes, after loading a hub you can user go to the My Data menu and paste in the location of a hub to display on any of the GenArk assembly hubs. The one special detail is that your Track Hub’s genomes.txt genomes line only needs to have the GCA/GCF number such as “genome GCF_001984765.1”. See this example hub.txt file for an idea of how a hub could be loaded on a GenArk hub. Here is a link that will load that hub on a GenArk hub for American beaver:
Can I share data on a GenArk assembly hub?
Yes, you can make a session and share the URL with others. Even better, publish your session to the Public Session page to make it more discoverable. See this previous blog about sharing data for more information: https://bit.ly/UCSC_blog_sharing
The next blog post in this series will provide some technical details about the GenArk hub architecture. The first post focussed on how to discover and access the hubs.
This entry written by Brian Lee. If after reading this blog post you have any public questions, please email genome@soe.ucsc.edu. All messages sent to that address are archived on a publicly accessible forum. If your question includes sensitive data, you may send it instead to genome-www@soe.ucsc.edu.
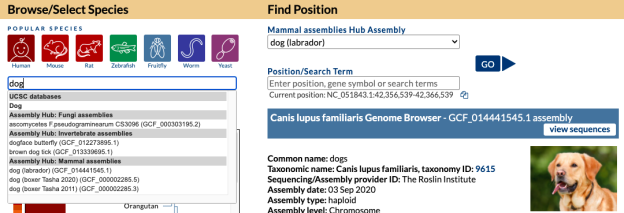

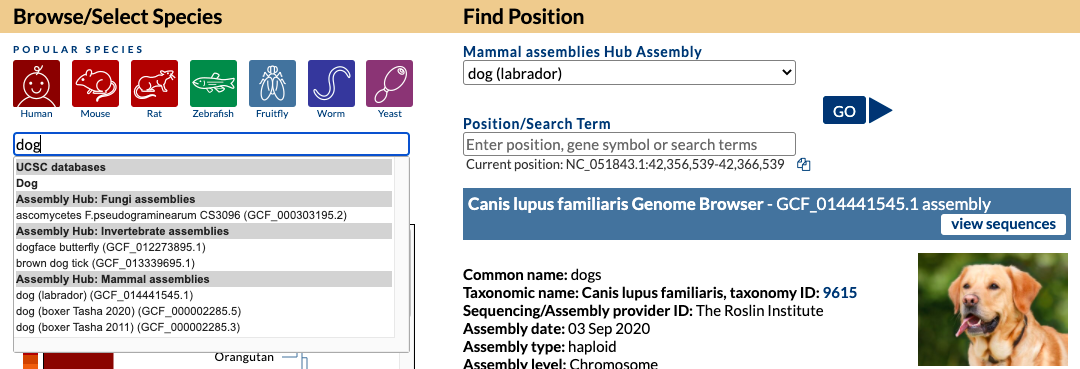 On the Gateway page in the top left box you can search a term such as “dog” and find all the genomes both hosted in our internal databases and in external Public Hubs that have dog in the name. In this image, a search for “dog” returns a top “Dog” match (UCSC database) as well as results for several species in Assembly Track Hubs that match on the term “dog” with the specific labrador dog breed selected from the GenArk Mammal Assemblies Hub (GCF_014441545.1).
On the Gateway page in the top left box you can search a term such as “dog” and find all the genomes both hosted in our internal databases and in external Public Hubs that have dog in the name. In this image, a search for “dog” returns a top “Dog” match (UCSC database) as well as results for several species in Assembly Track Hubs that match on the term “dog” with the specific labrador dog breed selected from the GenArk Mammal Assemblies Hub (GCF_014441545.1).