This blog post is the final of three to discuss the Genome Archive (GenArk) assembly hubs. This third post discusses the technical infrastructure of the GenArk hubs, while the first post was about accessing the data, and the second shared examples of using the data.
TECHNICAL DETAILS
What are the systems behind GenArk hubs?
In essence, GenArk hubs are assembly hubs that have been added as Public Hubs.
Anyone can build Track Hubs and Assembly Hubs, where finished hubs can then be requested to be published as Public Hubs. UCSC does have a Public Hub Guidelines page to encourage hub developers to document their data fully before submitting for inclusion, this is mainly to ensure users of Public Hubs know who to contact for their data and can understand what they are visualizing.
To aid independent groups that do not want to build assembly hubs, when the underlying assembly data is already available at NCBI, our engineers have crafted scripts to build these files automatically. GenArk scripts pull data from NCBI and then programmatically construct all of the binary-indexed files needed to visualize them on the UCSC Genome Browser. Additional special features have been included, especially the ability to generate and provide BLAT and PCR dynamic servers through the pre-generation of special index files. Our engineers have also optimized other elements of these GenArk Hubs by applying the latest available Track Hub features.
But what are the internal methods UCSC uses to populate the GenArk hubs?
Here at UCSC, an internal process maintains a local mirror image of the NCBI genome assembly resources. In essence, there is first a transfer of data with a rsync request, rsync://ftp.ncbi.nlm.nih.gov/genomes/all/GC[AF]/ to a matching local hierarchy of directories. These matching directory structures and naming conventions for files enable scripting procedures to automatically find and process the source files into the formats the UCSC Genome Browser recognizes to visualize data, mainly byte-range accessible binary-indexed versions of the data. A Perl script, doAssemblyHub.pl, manages all the steps of the procedure (https://genome-source.gi.ucsc.edu/gitlist/kent.git/blob/master/src/hg/utils/automation/doAssemblyHub.pl).
So how are gene tracks created for these GenArk hubs?
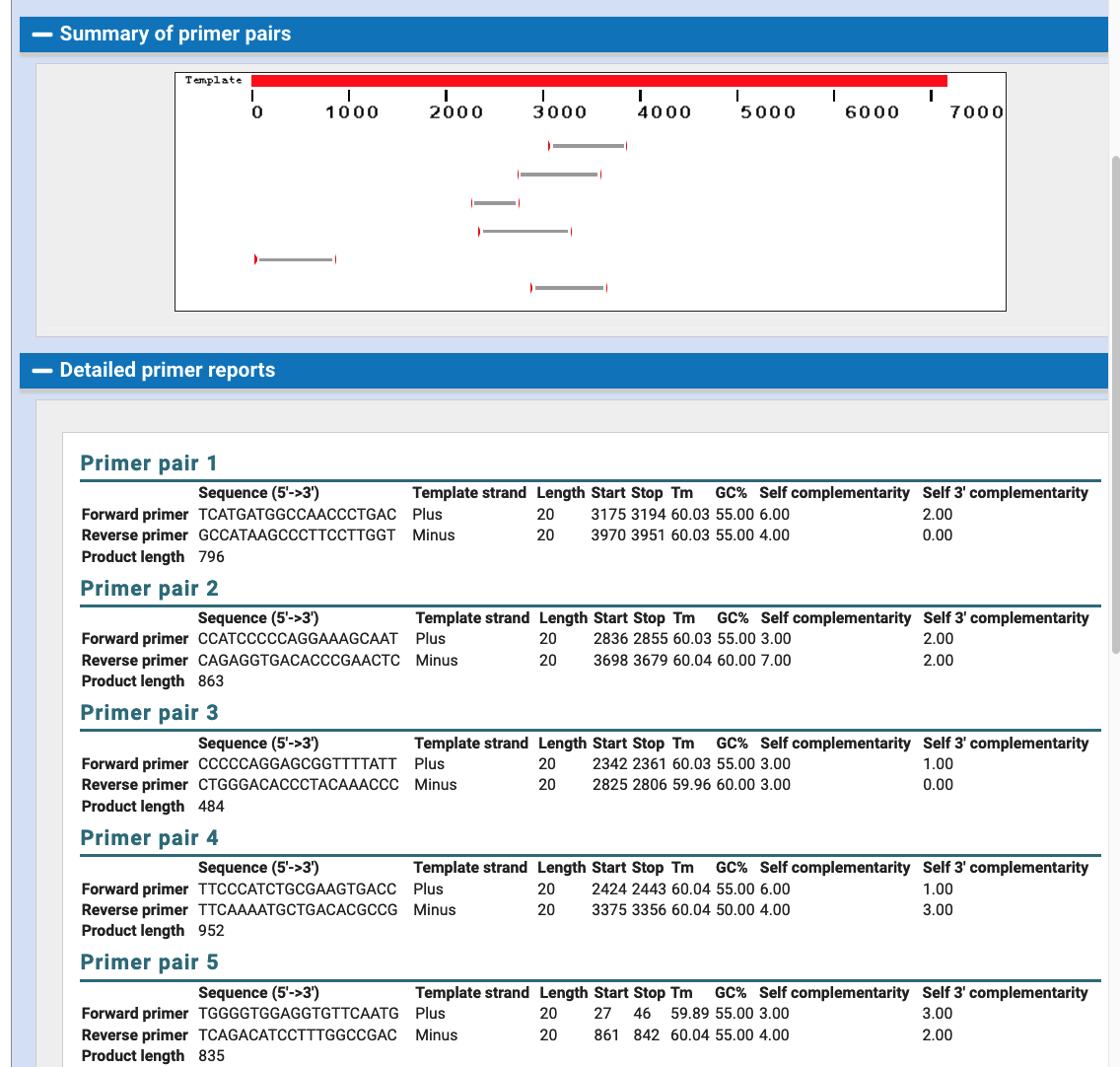
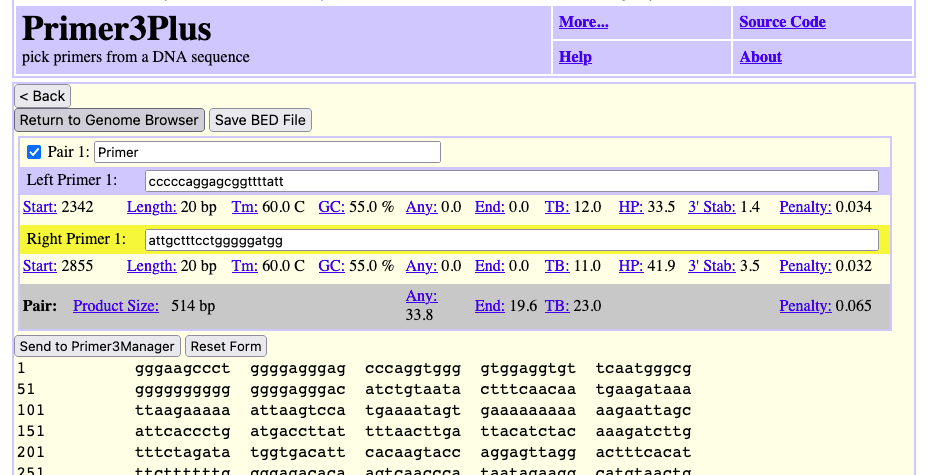
For the assemblies with GCF accessions, the script uses the supplied data of the NCBI gene annotations to create gene tracks that are provided with those files. A specific gene track type called bigGenePred, https://genome.ucsc.edu/goldenPath/help/bigGenePred.html, allows amino acid displays when zoomed-in at the base level. Likewise, a bigGenePred track is made to display Xeno RefGene data which is computed from a selection of best alignments of RefSeq mRNA sequences from many organisms to the genome, using the BLAT (http://www.kentinformatics.com/) algorithm (blat -noHead -q=rnax -t=dnax -mask=lower target.fa.gz query.fa.gz results.psl). Another bigGenePred track is made using the Augustus gene prediction software (http://bioinf.uni-greifswald.de/augustus/) from the Stanke lab.
How are the other GenArk annotation tracks made?
GenArk assemblies also have Repeat Masker tracks, which use the data when supplied from NCBI source. Otherwise, the track can be computed with a local installation of the Repeat Masker software (https://www.repeatmasker.org/). The Simple Repeats track is computed with the Tandem Repeats Finder software (https://tandem.bu.edu/trf/trf.submit.options.html) and the Window Masker track is computed with the WindowMasker software included in the NCBI C++ toolkit (https://ftp.ncbi.nih.gov/toolbox/ncbi_tools++/CURRENT/). The CpG Islands are computed with a modification of a program developed by G. Miklem and L. Hillier and the GC Percent track is computed using the ‘kent’ command hgGcPercent (http://hgdownload.soe.ucsc.edu/admin/exe/). Examining the doAssemblyHub.pl script (https://genome-source.gi.ucsc.edu/gitlist/kent.git/blob/master/src/hg/utils/automation/doAssemblyHub.pl), will illustrate more details about how individual steps are run (i.e., hgGcPercent -wigOut -doGaps -file=stdout -win=5 -verbose=0 test ../../\$asmId.2bit | gzip -c > \$asmId.wigVarStep.gz).
What if I don’t find my Assembly in the GenArk collection?
If you can’t find the assembly you want in the GenArk hub collection, but you do already have the GCA/GCF identifier you can email us at our public mailing-list genome@ucsc.soe.edu to request we add the assembly to the GenArk collection. This archived mailing-list is searchable from links on our contacts page, http://genome.ucsc.edu/contacts.html. Alternatively, if you don’t want your request to be public, you can email our private internal mailing-list at genome-www@soe.ucsc.edu. Also, since this original blog post, we created a new assembly request page, you can find details in this 4th GenArk blog post.
What if my assembly doesn’t have a GCA/GCF NCBI accession?
If NCBI does not have a GCA/GCF accession for your assembly then our scripts will not be able to pull the data and generate the GenArk hub. You will need to deposit the assembly at NCBI and notify us once the assembly has become available. You can find directions at NCBI for how to submit new genomes: https://www.ncbi.nlm.nih.gov/assembly/docs/submission/
A future manuscript is also in the works to further detail the background of the GenArk hubs.
This was the final blog in a three-part series about GenArk hubs authored by Brian Lee. The first post focused on how to discover and access the hubs, while the second blog post provided tutorial examples of using the GenArk hubs, such as the BLAT and PCR tools that are available, or how you can send DNA of any Assembly Hubs to External Tools for processing.
If after reading this blog post you have any public questions, please email genome@soe.ucsc.edu. All messages sent to that address are archived on a publicly accessible forum. If your question includes sensitive data, you may send it instead to genome-www@soe.ucsc.edu.
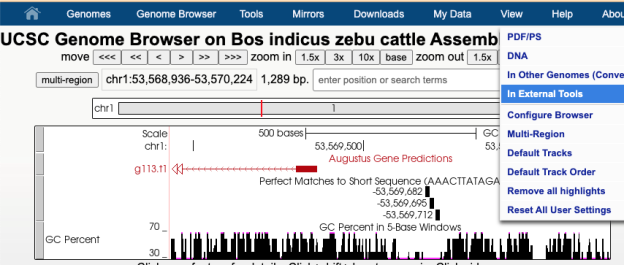
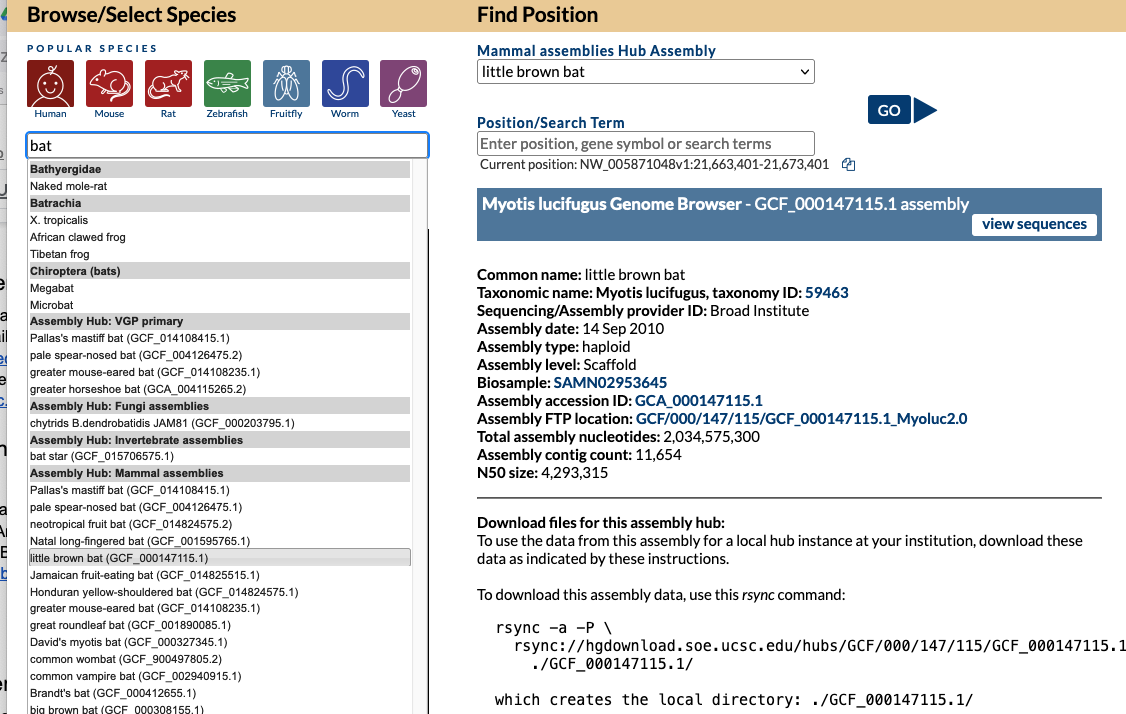

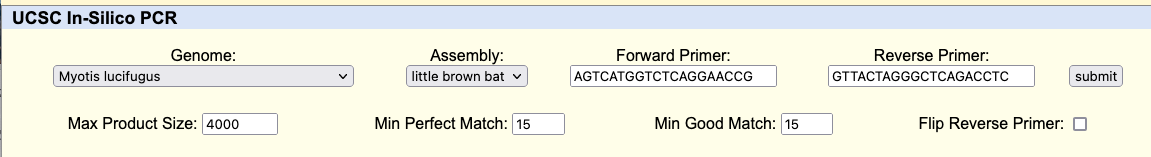
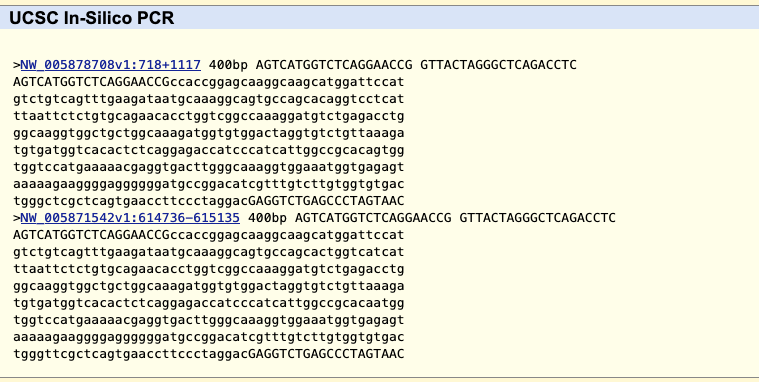
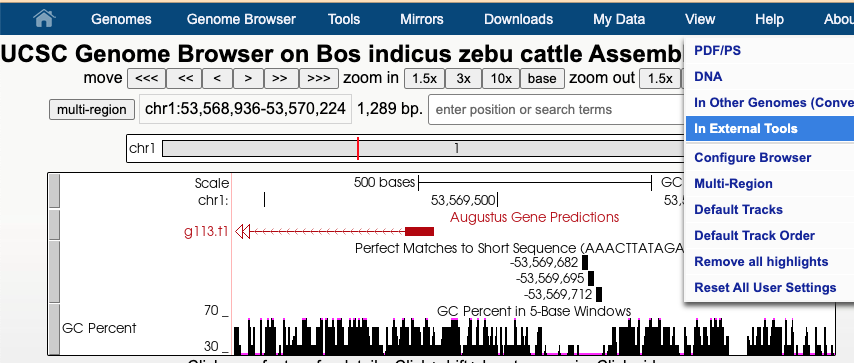
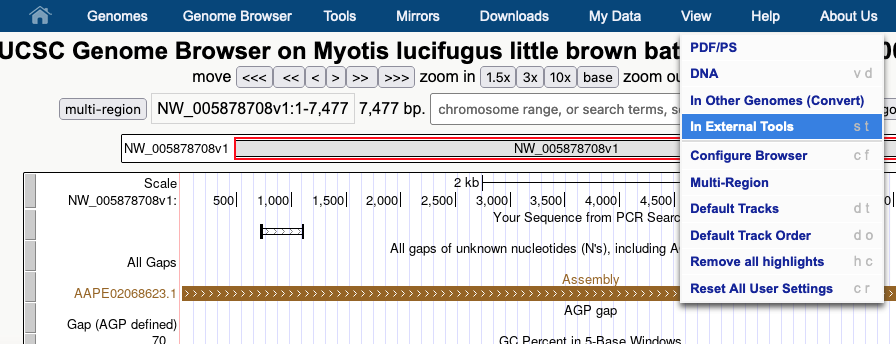 In this image a 7,477 bp region will be selected to be sent to external sites where selecting “In External Tools” under the View menu will result in a pop-up of various options.
In this image a 7,477 bp region will be selected to be sent to external sites where selecting “In External Tools” under the View menu will result in a pop-up of various options.